
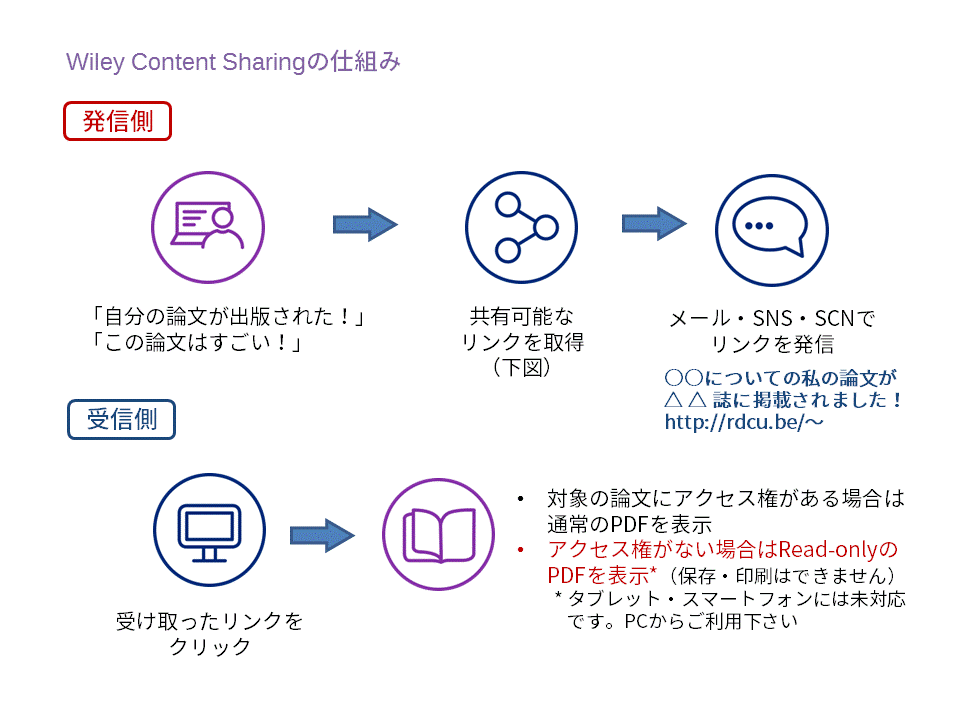
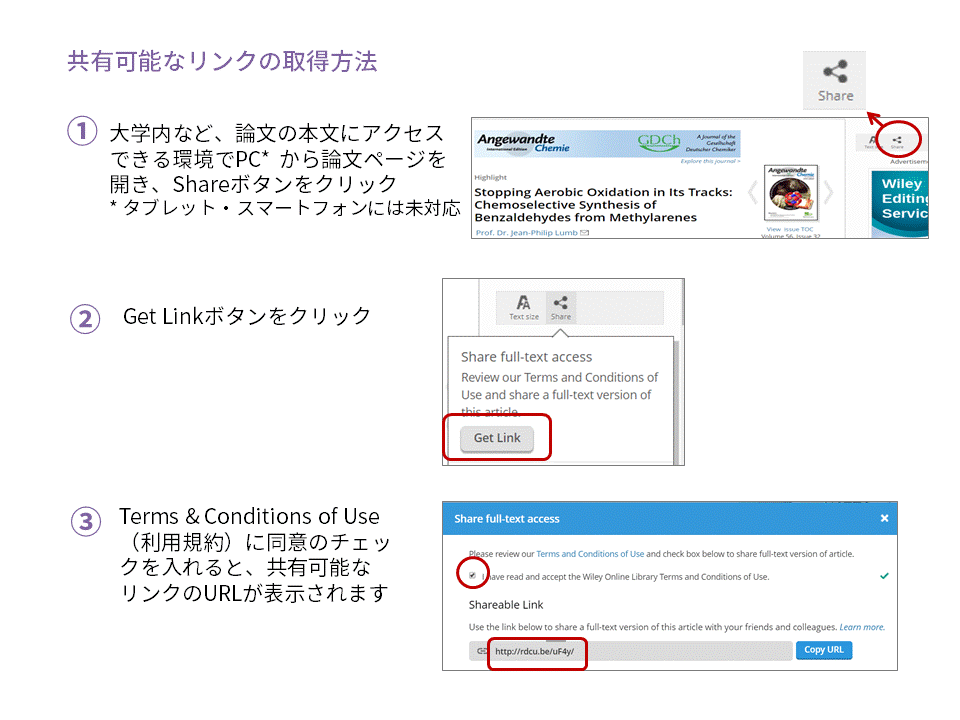
日本で8月に最もよく売れたWileyの医学・看護・獣医学書トップ5をご紹介します。タイトルまたは表紙画像をクリックすると、目次やサンプル章(Read an Excerpt)など、詳しい内容をご覧いただけます。
 1位 Shimizu’s Dermatology, 2nd Edition
1位 Shimizu’s Dermatology, 2nd Edition
Hiroshi Shimizu
ISBN: 978-1-118-78117-3
Paperback / 664 pages / December 2016
世界的な皮膚科医として知られる北海道大学・清水 宏教授の『あたらしい皮膚科学』は、皮膚科学の総合的な教科書として初版・第2版とも大成功を収めました。その初版を基にして、英語版 Shimizu’s Textbook of Dermatology が2007年に出版されましたが、今回刊行された本書は、新しい第2版(2011)をさらにアップデートした英語版で、旧版よりも内容が約20%増補されています。日本語版原著と同様に、最新の知識に基づいて皮膚科診療に必要な知識を漏らさず、しかも簡潔に分かりやすく解説しています。同時に、疾患名などの表記を、国際的に広く用いられる用語に改めています。精選された1,500点以上の画像も収録し、医学生から専門医まで、広く役立つ内容となっています。
 2位 TNM Classification of Malignant Tumours, 8th Edition
2位 TNM Classification of Malignant Tumours, 8th Edition
Edited by James D. Brierley, Mary K. Gospodarowicz, Christian Wittekind
ISBN: 978-1-119-26357-9
Paperback / 272 pages / December 2016
悪性腫瘍の病期分類に用いられ、現代のがん診療には欠かせない「TNM悪性腫瘍の分類」の、7年ぶりの改訂となる最新第8版です。TNM分類を定める国際組織Union for International Cancer Control (UICC)との全面的な提携によって編纂され、高い信頼性が保証されています。今回の改訂では、p16陽性中咽頭がん、胸腺がん、膵神経内分泌腫瘍、肉腫のそれぞれについて新たに病期分類が追加されました。がん診療に関わる人には必携の一冊です。
 3位 Manual of Small Animal Soft Tissue Surgery, 2nd Edition
3位 Manual of Small Animal Soft Tissue Surgery, 2nd Edition
Karen Tobias
ISBN : 978-1-119-11724-7
Hardcover / 624 pages / July 2017
幅広い小動物獣医師に役立つ軟組織外科手術マニュアルに、近年の新展開を取り入れた最新改訂版。押さえておくべき知識と有用な手技を、高画質のカラー写真とともにステップを追って伝えます。
 4位 Guide to Ruminant Anatomy: Dissection and Clinical Aspects
4位 Guide to Ruminant Anatomy: Dissection and Clinical Aspects
Mahmoud Mansour, Ray Wilhite, Joe Rowe
ISBN : 978-1-119-05102-2
Paperback / 296 pages / May 2017
代表的な反すう動物であるヤギとウシを対象にした、臨床上有用な解剖学ガイド。カラー図版244点を収録し、解剖学上の基礎を簡潔に解説します。章末問題に加え、購入者専用ウェブサイトでは解答と動画を参照できます。
 5位 Clinical Manual of Blood and Bone Marrow Transplantation
5位 Clinical Manual of Blood and Bone Marrow Transplantation
Edited by Syed A. Abutalib, Parameswaran Hari
ISBN: 978-1-119-09545-3
Paperback / 424 pages / April 2017
血液専門医とそれをめざす研修医を対象に、臨床現場で求められる知識を提供する実用本位のマニュアル書です。図表やフローチャートが多用され、必要な情報をすばやく得られます。
ご注文は最寄りの書店・ネット書店で承ります。
















") Wiley Asia Blog (Health Sciences)
Wiley Asia Blog (Health Sciences) ワイリー・サイエンスカフェ(理工学ブログ)
ワイリー・サイエンスカフェ(理工学ブログ) Neutrino WebShop(株式会社ニュートリノ)
Neutrino WebShop(株式会社ニュートリノ) オーヴィス株式会社
オーヴィス株式会社